We create digital experiences that inspire, transform and create action
Every digital branding experience, e-commerce system, software user interface, and web application your audience confronts exudes your business image. IONA provides a creative and interactive experience design that is meaningful and valuable to your audience and follows your business goals.
Digital Brand Strategy
Branding is about creativity, consistency and clarity through the various mediums. IONA’s process from strategy to implementation into collateral, print campaigns, online experiences and applications is recognized as an industry leader.
Online Experience
Creativity merged with technical design and an analytical approach to search engine optimization is the combination for successful online experiences. IONA creates award winning on-line experiences through the creation of intelligent design through the eyes of the target audience.
Custom Applications
Every client is different. Scalability and customization are IONA’s strong suites and what our clients expect. From custom content management system build-outs to custom B-to-B applications, IONA has decades of experience creating solutions that clients rely on for daily operation.
Solution Management
IONA is a long term partner for our clients. We continue to manage solutions through evolving technology, business strategy, and marketing campaigns through continued design, technology, and support management for on-line communications and solutions.

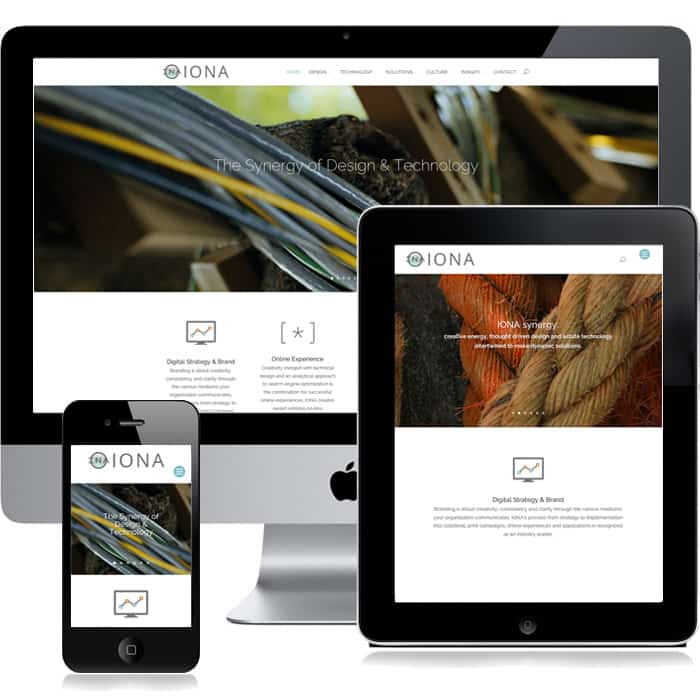
Responding to Users
Convenience and accessibility of any information your company communicates is the key to any businesses brand success. IONA has always been a stickler for reaching any and every user regardless of their high or low-level technology. Responsive design is simply the trendy term for what we have been doing for decades. Using some of the newest technology there is much to be inspired by. Learn More
- Digital Branding with Responsive Design 100%
- Leading Technology with Accessiblity for All 100%
- Continued Management with Strategy Planning 100%
Get Connected With IONA
Connect with IONA today to receive information about leading industry news and IONA services.